Perspectives
Biosimilars are here to help, really
Set biosimilars free to increase patient access to therapy by reducing costs
May 18, 2020If large employers had switched to biosimilars a couple of years ago, they could have already saved an average of $1.5 million each in drug spend.¹ This is according to a study reported in the emagazine, StatNews.
We’re pleased to report that Prime’s Blue Plan clients are starting to deliver those savings to employer groups.² But there is still much more to do.
Since 2015, the FDA has approved 24 biosimilars for nine reference biologics. But only 12 of those had been launched, as of the end of 2019. And those biosimilars had small market penetration.¹
- Why so few approvals?
- Why have so few of those few launched?
- And why the slow market uptake?
These questions will be covered in articles over the next couple of weeks.
Generics paved the way
Remember, before there were biosimilars, there were generics. In 1984, the Hatch-Waxman Amendments were passed to encourage prescription drug competition by providing a path for generic drug approvals.3,4 Drug companies applied to the U.S. Food and Drug Administration (FDA) with the abbreviated new drug application (ANDA). The ANDA showed that the generic drug’s active ingredient was same as the brand-name product’s.5
The FDA’s approval process required clinical studies showing that the generic drug performed the same way as the brand-name drug in the human body.
Once approved, the FDA certified that the generic drug is the same as the brand-name drug in dosage, form, safety, strength, route of administration, quality and performance.³
Generic drugs = brand-name drugs3,4
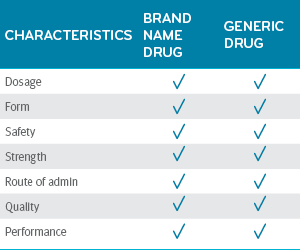 Generic drugs have produced tremendous savings for consumers: $293 billion in 2018 alone. (the last year for which numbers are available). More than 90 percent of all prescriptions filled in the United States are generic drugs. Yet these generic prescriptions amount to only 22 percent of drug costs.6
Generic drugs have produced tremendous savings for consumers: $293 billion in 2018 alone. (the last year for which numbers are available). More than 90 percent of all prescriptions filled in the United States are generic drugs. Yet these generic prescriptions amount to only 22 percent of drug costs.6
These savings didn’t happen all at once. In fact, it started out kind of slow, for reasons that will be in the next article.
Comparing generics and biosimilars
Generics are to brand-name drugs, what biosimilars are to biologics.
Almost. Biosimilars don’t have quite the same relationship to biologics that generic drugs have to brand-name drugs. But biosimilars are still an important way for improving patient access to specialty drugs and reducing costs.7
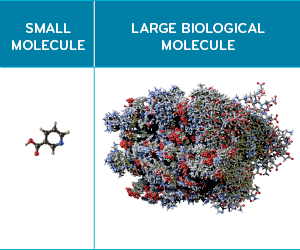 The generic drugs that launched in the ‘90s and ‘00s were all small-molecule drugs. These drugs all have small, basic molecular structures. Their chemical make-up is compact. They have a simple mechanism of action, and the manufacturing process is straightforward; few steps are needed.7
The generic drugs that launched in the ‘90s and ‘00s were all small-molecule drugs. These drugs all have small, basic molecular structures. Their chemical make-up is compact. They have a simple mechanism of action, and the manufacturing process is straightforward; few steps are needed.7
Biologics are large molecule drugs. Biologics can be thousands of times larger and have complex mechanisms of action. They are living cells or complex proteins, and are often genetically modified in some way. The biologic manufacturing process is elaborate and multi-faceted.7, 8 Relatively speaking, generics were much easier to produce.
25 years after generics, biosimilars get their own Act
The Biologics Price Competition and Innovation Act (BPCIA) of 2009 defined the biosimilar category of drugs.8 Like the Hatch-Waxman Amendments, it created a passing lane on the approval’s roadmap; this time for biosimilars. Drug manufacturers use the abbreviated biologic license application (ABLA) pathway for a biosimilar that is similar to a biologic reference product. Similar. It won’t be identical, like a generic. But similar still gets the job done.
Biosimilars deliver the same quality as the biologic reference product.5 Between a biosimilar and a biologic reference product, there are no clinically meaningful differences in dosage, form, safety, strength, route of administration, purity or potency.
Biosimilars and their reference products: No clinically meaningful difference
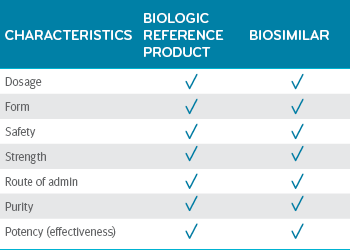 It’s in the name. Biosimilar. It’s similar, but not equivalent. It won’t be identical. The large molecule is too complex. An ABLA must include data that shows biosimilarity to the reference product. The manufacturer provides the comparison data that demonstrates its biosimilarity. The manufacturer prepares this data in a stepped approach, as described by an FDA guidance document.9,10,11 The FDA may ask for more data on a case-by-case basis before approving an ABLA. (In the generic ANDA, the paperwork is specific and finite.)
It’s in the name. Biosimilar. It’s similar, but not equivalent. It won’t be identical. The large molecule is too complex. An ABLA must include data that shows biosimilarity to the reference product. The manufacturer provides the comparison data that demonstrates its biosimilarity. The manufacturer prepares this data in a stepped approach, as described by an FDA guidance document.9,10,11 The FDA may ask for more data on a case-by-case basis before approving an ABLA. (In the generic ANDA, the paperwork is specific and finite.)
Not just one, but TWO kinds of biosimilars
The BPCIA also named a second pathway, for interchangeable biosimilars. The dual pathways – biosimilars and interchangeable biosimilars – have caused some confusion.
An interchangeable product has a much higher bar. It is expected to produce the same clinical result as the reference product in any given patient. Also, for products administered to a patient more than once, the risk in terms of safety and reduced efficacy of switching back and forth between an interchangeable product and a reference product will have been evaluated.
Then, an interchangeable biosimilar product may be substituted for the reference product without the involvement of the prescriber.
For this interchangeable biosimilar pathway, the FDA can ask for yet again even more additional studies focusing on how the drug moves through and affects the body. Interestingly, the FDA has not provided guidelines on the technology or documentation needed to demonstrate a biologic is interchangeable.12,13
Agencies and governments agree, that demonstrating an interchangeable biosimilar is beyond today’s technology.13 There is a marked reluctance to attempt to apply for an approval within this biosimilar category.
Appropriately, the marketplace currently has its focus on the first biosimilar pathway.
Biosimilars are just beginning to make an impact
The 12 biosimilars launched in the U.S. represent just a fraction of the potential biosimilars market. The market has been cautious, looking for more regulatory guidance and leadership. The FDA’s Biosimilar Action Plan of 2018 had many sound elements, but progress is slow. Forward motion gets bogged down by more urgent, systemic health care issues.
Prime provides its clients with strategic information about the drug pipeline on an ongoing basis. Prime’s Medical Business Committee involves clients in the decision-making process on new specialty medicines, including biosimilars. It’s at those meetings where we have made multi-million dollar biosimilar recommendations for clients.
References
- Large employers could have saved money if more biosimilars had been prescribed, By ED SILVERMAN, April 2, 2020. STAT NEWS. Accessed at: https://www.statnews.com/pharmalot/2020/04/02/biosimilars-biologics-drug-prices-
- https://www.primetherapeutics.com/en/news/prime-insights/2020-insights/insights-2020-never-give-up-on-biosimilars.html
- Drug Price Competition and Patent Term Restoration Act of 1984. 21 U.S.C. § 355 (1984).
- Grabowski H, Long G, Mortimer R, Boyo A. Updated trends in US brand-name and generic drug competition. J Med Econ. 2016;19(9):836-844. doi: 10.1080/13696998.2016.1176578.
- Abbreviated new drug application (ANDA). FDA. Updated May 22, 2019. Accessed November 14, 2019. www.fda.gov/drugs/types-applications/abbreviated-new-drug-application-anda.
- The Case for Competition: 2019 Generic Drug & Biosimilars Access & Savings in the U.S. Report, Association for Affordable Medicines. Accessed at: https://accessiblemeds.org/sites/default/files/2019-09/AAM-2019-Generic-Biosimilars-Access-and-Savings-US-Report-WEB.pdf
- Generic drug facts. FDA website. Updated June 1, 2018. Accessed November 13, 2019.www.fda.gov/drugs/generic-drugs/generic-drug-facts.
- What is a biosimilar? U.S. FDA. Accessed at: https://www.fda.gov/media/108905/download
- FUTURE ONCOLOGY. VOL. 15, NO. 3. REVIEW Comparative immunogenicity assessment of biosimilars. Thomas Schreitmüller, Bettina Barton, Artem Kharkov & Georgios Baals. Published Online:3 Oct 2018. Accessed at: https://doi.org/10.2217/fon-2018-0553https://www.futuremedicine.com/doi/full/10.2217/fon-2018-0553
- Gazania AA, Baggio G, Caputi AP, et al. Biosimilar drugs: concerns and opportunities. Biodrugs. 2007;21:351-356. Accessed at: https://www.ncbi.nlm.nih.gov/pubmed/18020619
- Mulcahy AW, Hlávka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Published 2017. RAND Corporation website. Accessed at: rand.org/content/dam/rand/pubs/perspectives/PE200/PE264/RAND_PE264.pdf
- Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. FDA website. Published April 2015. Accessed November 14, 2019. www.fda.gov/media/82647/download.
- Report: Emerging Health Care Issues: Follow-on Biologic Drug Competition. Federal Trade Commission Report, June 2009. Ftc.gov.
Related news
Perspectives
July 25, 2024
Quarterly Drug Pipeline: July 2024
Clinical insights and competitive intelligence on anticipated drugs in development
Perspectives
July 22, 2024
Oncology Insights: 2024 ASCO Annual Meeting key findings
Findings from this year’s American Society of Clinical Oncology (ASCO) Annual Meeting will likely lead to clinical practice changes and U.S. Food and Drug Administration (FDA) drug approvals or expansions
Perspectives
July 16, 2024
LISTEN NOW: Beyond the business – Stories of corporate kindness | Pharmacy Friends Podcast
In this episode, we talk about how our employees' help goes beyond our work in health care, aiding in philanthropic efforts